Introduction
Increasing evidences unequivocally demonstrate that beta-lapachone (β-Lap) is cytotoxic to cancer cells expressing NAD(P)H:quinone oxidoreductase (NQO1) [1-18]. Importantly, the level of NQO1 activity in many cancers is significantly higher than that in normal tissues indicating that cancer cells are more vulnerable to β-Lap than normal cells. Recent studies clearly demonstrated that β-Lap is not only cytotoxic by itself but it also potentiates the response of cancer cells to ionizing radiation (IR) [3-6], chemotherapy [7], and hyperthermia [8-11]. The purpose of this article is to review recent reports on the molecular mechanisms underlying the anti-cancer effect of β-Lap alone or in combination with ionizing radiation.
β-Lapachone
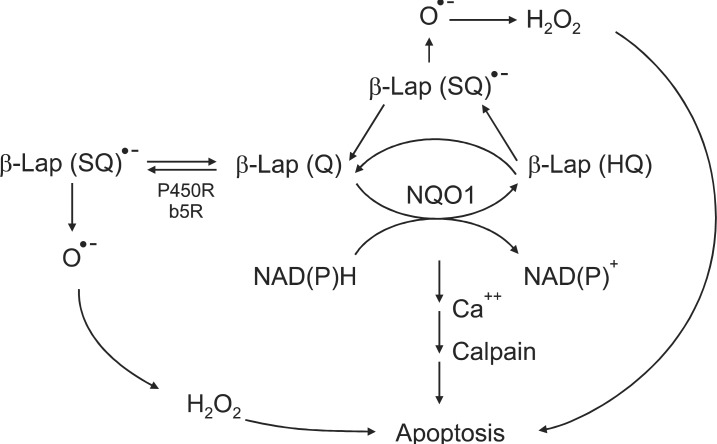
β-Lap was originally obtained from Lapacho trees, which are abundant in South America. Since ancient times, the leaves and bark of Lapacho tree have been used as tea and herbal medicine for various diseases. β-Lap is now chemically synthesized or prepared in quantities from natural lapachol. β-Lap exerts a divergent therapeutic activities such as 1) anti-bacterial and anti-fungal effect, 2) anti-leukemia virus effect, 3) anti-HIV-virus effect, 4) anti-inflammation effect, 5) anti-psoriasis effect, and 6) anti-cancer effect [1,2]. In recent years, the pharmacological effects of β-Lap have attracted considerable attention particularly in the cancer research community. β-Lap has been shown to induce apoptosis in various types of cancer cell lines including prostate, lung, pancreas, colon, ovary and breast cancer [1-18]. Furthermore, β-Lap has recently been demonstrated to synergistically interact with IR [3-6] and certain chemotherapy drugs [7] and heat shock [8-11] to kill cancer cells. There have been extensive investigations on the mechanisms of the anti-cancer effects of β-Lap. Cell death caused by β-Lap was initially attributed to β-Lap-induced inhibition of topoisomerase I activation [16,19-22] and alteration in topoisomerase II activities [23,24]. However, subsequent studies indicated that the β-Lap-induced alteration in topoisomerase activities observed in cell free system in vitro may not be the major player for β-Lap-induced cell death in vivo [16]. Other studies indicated that β-Lap induces apoptosis by interrupting cell cycle progression and that the synergistic interaction of β-Lap and other cytotoxic agents such as Taxol is caused by inhibition of multiple cell cycle checkpoints [17-18,25]. Further studies have indicated that futile cycling between oxidized quinone form and two-electron reduced hydroquinone (HQ) form of β-Lap mediated by NAD(P)H:quinone oxidoreductase (NQO1) is the cardinal process for the killing of cancer cells by β-Lap (Fig. 1).
NAD(P)H:Quinone Oxidoreductase
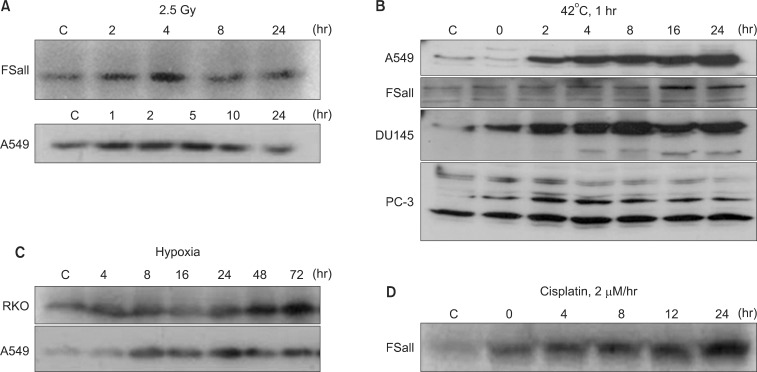
NQO1, also called DT-diaphorase (EC 1.6.99.2), is an obligatory two-electron reductase [26-36]. This flavoprotein enzyme converts quinone compounds in cells to HQ through two-electron reduction using NADH or NAD(P)H as electron sources (Fig. 1). The hydroquinones are then conjugated with glutathione, glucuronic acid or other moieties and removed from cells [33,34]. In a sense, NQO1 competes with NADPH-cytochrome p-450 reductase (P450R) and NADH-cytochrome b5 reductase (b5R), which all causes one electron reduction of quinones to semiquinones [28,37-39]. Unlike the hydroquinone, the semiquinones induce redox cycling and generate reactive oxygen species (ROS), thereby causing oxidative damages to cells. Therefore, the two-electron reduction of quinones to relatively stable hydroquinone by NQO1 has been suggested to be a detoxification process, and thus NQO1 is regarded as a chemo-preventive enzyme. In this connection, the chemo-preventive activity of a variety of synthetic compounds and also dimethyl maleate (DMM) or dimethyl fumarate (DMF), which are found in vegetables and fruits, have been attributed to their ability to upregulate NQO1 and other phase II detoxifying enzymes in cells [26]. Contrary to above, certain quinone-containing compounds become powerful anti-cancer drugs when they are two-electron reduced by mediation of NQO1. For example, NQO1 bioactivates mitomycin C (MMC) to a hydroquinone form, which is a far more potent alkalizing agent than the parent MMC [37-39]. NQO1 is ubiquitously expressed, but the levels of NQO1 in various human malignant tumors are significantly higher than that in adjacent normal tissues. For example, the NQO1 gene level in human hepatocarcinomas and hepatoblastomas is respectively 20-50 fold and 50-fold higher than that in normal liver tissues [40-42]. Interestingly, as mentioned above, DMM, DMF, fish oil, 13-cis-retinoic acid, aspirin, and ibuprofen have been reported to upregulate NQO1 [26]. Importantly, various anti-cancer agents such as IR [3-6], chemotherapy drugs [7] and hyperthermia (heat shock) [8-11] and hypoxia (our unpublished observation) also increase NQO1 levels (Fig. 2). The fact that hypoxia upregulates NQO1 has a significant clinical implication because intratumor environment is intrinsically hypoxic, which may lead to upregulation of NQO1 in cancer cells. The mechanisms by which NQO1 expression is transcriptionally regulated by aforementioned factors have been investigated [10]. Several essential cis-elements, including an antioxidant response element (ARE), have been identified within the human NQO1 gene promoter region [26,43]. The ARE element, which is comprised of several nuclear transcription factors, is responsible for basal expression as well as induction of NQO1 not only by antioxidants but also by oxidants and xenobiotics [44]. It is highly probable that IR, heat shock, hypoxia, acidic environment and certain chemotherapy drugs activate NQO1 gene through similar mechanisms.
Role of NQO1 in Anti-Cancer Effect of Bioreductive Drugs
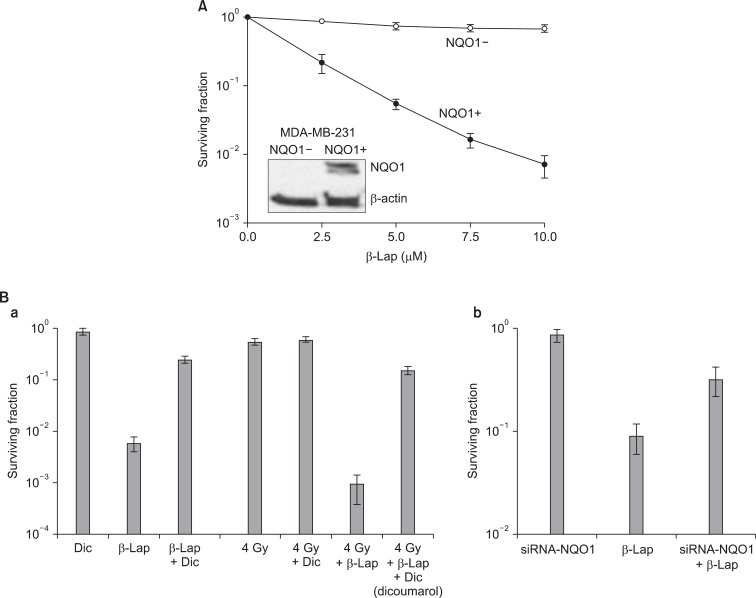
As mentioned in the preceding sections, recent experimental results by us as well as others have indicated that two-electron reduction of β-Lap mediated by NQO1 may be the initial step of the β-Lap-induced cell death pathway. In support of this view, the cellular sensitivity to β-Lap has been observed to be closely related to the level of NQO1 expression [12,26,45,46]. In addition, NQO1 inhibitors such as dicoumarol [13,30,45] effectively inhibit β-Lap-induced cell death. Furthermore, stable transfection of NQO1 expression plasmid into cells lacking NQO1 increased the sensitivity of cells to β-Lap [10,12] (Fig. 3). It has been observed that 1 mol of β-Lap could oxidize as many as 10 mol of NADH in 10 seconds or 100 mol of NADH in 10 minutes. Unlike the two-electron reduction products of other quinones, the hydroquinone form of β-Lap is unstable and rapidly undergoes autoxidation to the parent quinone [13]. It is believed that NQO1 induces "futile cycling" between quinone and hydroquinone forms of β-Lap utilizing NADH and NAD(P)H as electron sources. Consequently, NADH and NAD(P)H are severely depleted and Ca2+ is released from the endoplasmic reticulum into the cytosol after β-Lap treatment [12-15]. The increase in cytosolic Ca2+ is followed by depletion of ATP, depolarization of mitochondrial membrane, release of cytochrome C, and activation of Ca2+- dependent calpain or similar proteases. The unique features of apoptosis caused by β-Lap treatment is the degradation of p53 protein to a 40 kDa fragment and the cleavage of poly (ADP-ribose) polymerase (PARP) to a 60 kDa fragment instead of 89 kDa fragment usually observed in caspase-mediated apoptosis. The cleavage of p53 and PARP appeared to be caused by Ca2+ dependent calpain. It was therefore proposed that β-Lap activates calpain-like proteases, thereby degrading vital proteins in the cells leading to caspase-independent apoptosis [12-15] (Fig. 1). In addition to the aforementioned molecular changes caused by depletion of NADH and NAD(P)H, generation of ROS may also be involved in β-Lap-induced cell death. It is likely that β-Lap (HQ), a hydroquinone formed as a result of two-electron reduction of β-Lap, may not directly oxidize back to β-Lap. Instead, β-Lap (HQ) may be oxidized first to a one-electron reduced intermediate, i.e., semiquinone β-Lap (SQ)·- in a manner analogous to the oxidation of the hydroquinone form of MMC to the one-electron reduced form of MMC [13,30,37,46-50] (Fig. 1). The active semiquinone β-Lap (SQ)·- may then induce ROS and redox cycling and generate superoxides and H2O2, thereby triggering caspase-dependent apoptosis. In support of this notion, β-Lap has indeed been shown to induce caspase-dependent apoptosis and cleavage of PARP to an 89 kDa fragment under certain conditions [14]. In the cells lacking functional NQO1, such as HL-60 cells, β-Lap induces caspase-dependent apoptosis by generating ROS [50].
Cell Death by β-Lap Alone
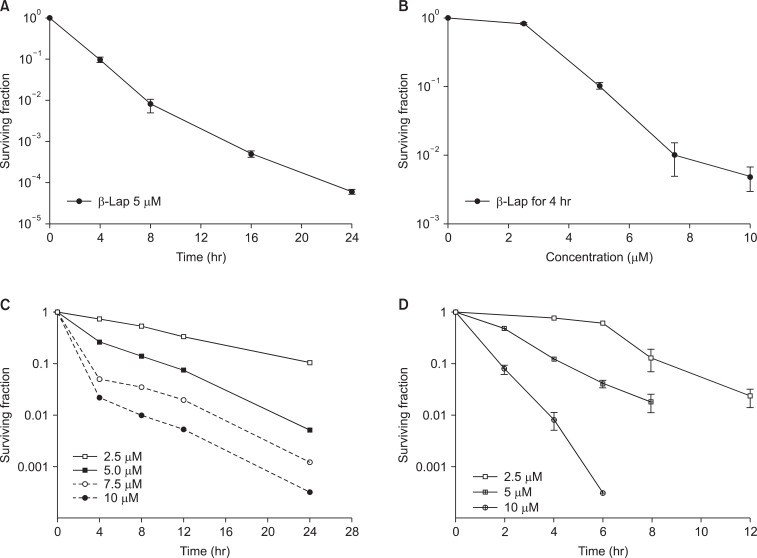
The cytotoxic effects of β-Lap alone have been investigated with a variety of cell lines. Fig. 4 shows the β-Lap-induced clonogenic deaths of A549 human lung epithelial cancer cells (A, B), PC3 human prostate cancer cells (C; our unpublished observation), and FSaII mouse fibrosarcoma (D). It is shown that incubation with 5 µM β-Lap for 4 hours at 37℃ reduced the clonogenic survival to about 10% in most of the cell lines studied. The importance of the role of NQO1 in the β-Lap-induced cell killing is clearly demonstrated in Fig. 3A, which shows that the β-Lap sensitivity of human MDA-MB-231 breast cancer cells deficient of NQO1 (NQO1-) and that of MDA-MB-231 cells stably transfected with ngo1 (NQO1+) (10). Incubation with β-Lap caused dose dependent clonogenic death in MDA-MB-231 (NQO1+) cells whereas it caused little death in MDA-MB-231 (NQO1-). Dicoumarol is an inhibitor of NQO1 and treatment of cells with dicoumarol significantly protected cancer cells from β-Lap-induced death in various cell lines [3,5-8]. In agreement with these previous reports, Choi et al. [6] observed that dicoumarol significantly protected A549 human lung cancer cells from β-Lap-induced clonogenic death (Fig. 3B-a). Furthermore, suppression of NQO1 activity in A549 (NQO1+) cells by transfecting the cells with siRNA-NQO1 rendered the cells resistant to β-Lap (Fig. 3B-b). Collectively, these results clearly demonstrated that NQO1 plays a crucial role for the cell death caused by β-Lap. The mechanisms by which NQO1 mediates β-Lap-induced cells death has already been discussed in the preceding sections. Note that, in addition to promoting futile cycling between quinone and hydroquinone forms of β-Lap, NQO1 may also be involved in other molecular and cellular changes caused by β-Lap such as cell cycle progression by affecting p53, p21 and cell cycle checkpoint as described in following sections.
Cell Death by β-Lap in Combination with Irradiation
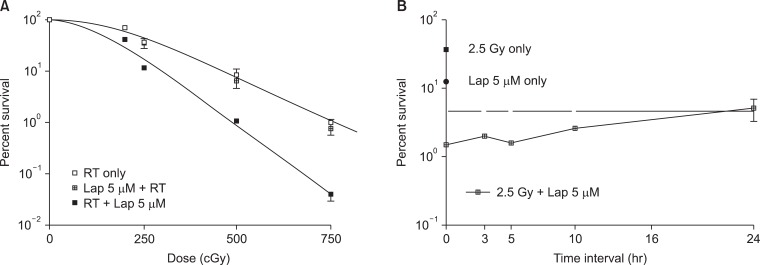
It was previously reported that β-Lap increases the radioresponse of cells by inhibiting the repair of radiation-induced potentially lethal damage (PLD) [1,19]. Several subsequent reports demonstrated that β-Lap and IR interact in synergistic manner in killing cancer cells. Park et al. [3] reported that treatment of FSaII fibrosarcoma cells of C3H mouse with β-Lap prior to IR exposure caused little change in the response of the cells to irradiation. On the other hand, the radiation survival curves of the cells irradiated first and then immediately treated with β-Lap was much steeper than that of the cells exposed to IR alone (Fig. 5A). Importantly, as shown in Fig. 5B, β-Lap treatment applied 3-5 hours after irradiation was as effective as that applied immediately after irradiating the cells to increase the radiation-induced cell death. It was apparent that the combination of irradiation followed by β-Lap treatment was synergistic even when cells were treated with β-Lap as late as 10 hours after irradiation. As shown in Fig. 2, the NQO1 level in FSaII cells markedly increased for 24 hours after 2.5 Gy irradiation. The authors therefore concluded that irradiation enhanced the response of tumor cells to β-Lap by increasing NQO1 activity. Similar observation was reported by Choi et al. [6] who studied the combined effect of irradiation and β-Lap using A549 lung cancer cells. The NQO1 level in the A549 cells was elevated for 24 hours after irradiation with 4 Gy and the irradiated cells were markedly sensitive to β-Lap for as long as 24 hours after irradiation. Essentially similar effect was obtained in a study with DU-145 prostate cancer cells that irradiation increased NQO1 level, thereby it sensitized the cells to subsequent treatment with β-Lap [5]. The notion that the post-irradiation increase in the response of cells to β-Lap is due to radiation-induced elevation of NQO1 was further supported by the observations that transfection of cells with NQO1-siRNA [6] or dicoumarol treatment [3,5] significantly inhibited the post-irradiation sensitization of cancer cells to β-Lap.
However, further studies revealed that the synergistic effect of irradiation and β-Lap to kill cancer cells is due not only to radiation-induced sensitization of cells to β-Lap, but also to the inhibition of sublethal radiation damage repair by β-Lap. It is well known that irradiation in two divided doses is less effective than that in a single dose with identical total dose because the sublethal radiation damage caused by the first dose is repaired by the time when the second irradiation is delivered. To investigate whether β-Lap affects the repair of sublethal radiation damage, cells were maintained in culture media containing β-Lap during the interval between the two fractionated irradiation and the clonogenic survival was determined [3,5]. It was clear that β-Lap suppressed the repair of sublethal radiation damage that take place during the interval of two fractionated irradiation. We may conclude that the synergistic interaction between irradiation and β-Lap in killing cancer cells results from two distinct mechanisms: 1) irradiation increases the NQO1 activity, thereby it sensitizes cells to β-Lap, and 2) β-Lap suppresses the repair of sublethal radiation damage. The effectiveness of β-Lap in combination with radiotherapy in vivo has been studied. The suppression of tumor growth caused by β-Lap in combination with a single dose irradiation was significantly greater than that caused by β-Lap or irradiation alone in several animal tumor models [3,4,6].
Effects of β-Lap Alone or in Combination with Irradiation on Cell Cycle Progression
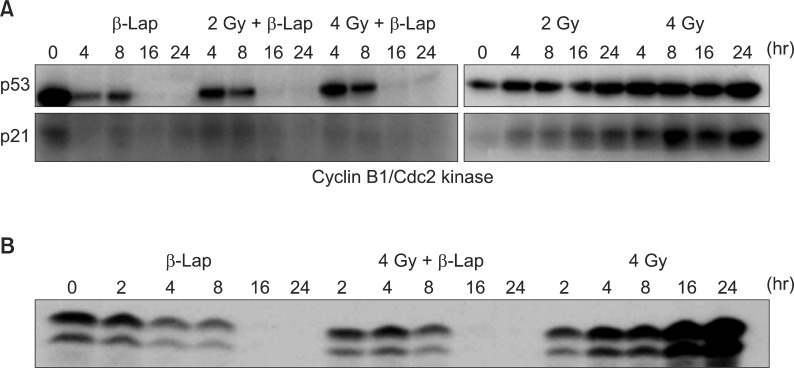
β-Lap has been reported to kill cancer cells by interrupting cell cycle progression, thereby causing apoptosis [1,17,18]. We have elucidated the effects of β-Lap on various molecular signals known to be involved in cell cycle progression and apoptosis [4,6]. Interestingly, the expressions of p53 and p21, which are intimately involved in cell cycle progression and apoptosis, were markedly suppressed within 24 hours after cells were treated with β-Lap for 4 hours (Fig. 6). In view of the previous report that p53 is stabilized by NQO1 [4,6], it is highly probable that depletion of NQO1 leads to disintegration of p53 molecule. We may then hypothesize that β-Lap, which is a substrate of NQO1, competes with p53 for NQO1and interrupts the NQO1-mediated stabilization of p53. It is likely that the decline in p21 activity resulted from the marked decline in p53. Interestingly, when irradiated cells were treated with β-Lap, the expression of p53 and p21 was similar to that as in the cells treated with β-Lap alone, demonstrating that β-Lap nullified the effect of radiation to increase the expression of p53 and p21. Cyclin B1/cdc2 kinase is known to promote the progression of G2 cells to mitosis. It is a well known fact that irradiation causes G2 arrest and increases cyclin B1/cdc2 kinase activity. Kim et al. [4] reported that an increase in the G2 cell population after irradiation increased the cyclin B1/cdc2 kinase activity, which in turn, accelerated the exit of cells from the G2 phase to mitosis and that the β-Lap completely prevented the radiation-induced G2 arrest as well as the radiation-induced upregulation of cyclin B1/ldc2 kinase activity. It is tempting to conclude that the progressive decline in the G2 cell population after β-Lap treatment removed the necessity for an increase in the cyclin B1/cdc2 kinase activity in irradiated cells. The mechanisms underlying the suppression of p53 activity and that in cyclin B1/cdc2 kinase activity by β-Lap in the irradiated cells remain to be investigated. Nevertheless, whatever the underlying mechanisms might be, it would be reasonable to speculate that the suppression of radiation-induced increase in p53 expression and increase in cyclin B1/cdc2 kinase activity and radiation-induced G2 arrest by β-Lap are related to the synergistic effects of IR and β-Lap against cancer cells.